SEARCH
 = Registered users
= Registered users = Paid-up subscribers
= Paid-up subscribersNairn C, Ralston SH. Diagnosis and management of Paget’s disease of bone. Practitioner November 2020;264(1842):11-15
Diagnosis and management of Paget’s disease of bone
24 Nov 2020
AUTHORS
Dr Catherine Nairn MB ChB, GP trainee, St Triduana’s Medical Practice, Edinburgh, UK
Professor Stuart H Ralston MB ChB MD FRCP FMedSci FRSE FFPM (Hon), Professor of Rheumatology, Centre for Genomic and Experimental Medicine, MRC Institute of Genetics and Molecular Medicine, University of Edinburgh, Edinburgh, UK
Competing interests: Professor Stuart Ralston is Chairman of the Board of Trustees of the Paget’s Association. He has received research grant funding from the European Research Council for research into Paget’s disease and from the Efficacy and Mechanism Evaluation programme of the National Institute for Health Research for the ZiPP study. Novartis supplied zoledronic acid and placebo infusions for this study. infusions for this study. Dr Catherine Nairn has no competing interests
Article
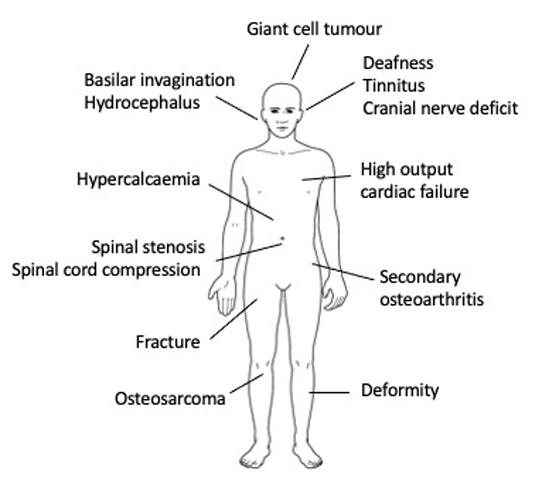
Paget’s disease of bone is a metabolic disease in which focal abnormalities of bone remodelling occur in one or more skeletal sites. The affected bones enlarge and may become deformed and this can lead to complications including bone pain, pathological fractures, secondary osteoarthritis, deafness and nerve compression syndromes. Paget’s disease is seldom diagnosed below the age of 50 but the incidence doubles every 10 years thereafter to 5-7/10,000 population per year by the ninth decade in the UK. Many people with Paget’s disease do not experience symptoms and it is estimated that only 7-15% of people with X-ray evidence of Paget’s disease come to medical attention. The three main risk factors are age, male gender and family history. People with a first-degree relative with Paget’s disease have a seven-fold increased risk of developing the disease. The most common presenting features in those that do present clinically are bone pain (52.2%), bone deformity (21.5%), deafness (8.9%) and pathological fractures (8.5%). Rarely, Paget’s disease may present with osteosarcoma (0.3% of patients). A radionuclide bone scan is the most sensitive diagnostic investigation. If this is unavailable, plain X-rays of the abdomen (including the ribs and femoral heads), both tibias and the skull (including facial bones) will detect 93% of cases. Patients who have bone deformity or symptoms that might be due to Paget’s disease should be referred to secondary care. The main indication for medical treatment is bone pain localised to an affected site where there is evidence of increased metabolic activity. Bisphosphonates are the treatment of choice.
Paget’s disease of bone is a metabolic disease in which focal abnormalities of bone remodelling occur in one or more skeletal sites.1
It is seldom diagnosed below the age of 50 but doubles in incidence every 10 years thereafter to affect 5-7/10,000 population per year by the ninth decade in the UK. Men are more often affected than women in a ratio of 1.4:1.2
Under normal circumstances, bone is renewed and repaired throughout life in an orderly fashion through bone remodelling. In Paget’s disease, the bone remodelling process is accelerated due to an increase in osteoclastic bone resorption coupled with increased and disorganised bone formation.
The affected bones enlarge and may become deformed and this can lead to complications including bone pain, pathological fractures, secondary osteoarthritis, deafness and nerve compression syndromes.2 Osteosarcoma is a rare complication which is thought to arise as the result of dysregulated osteoblast activity.
Risk factors
The three main risk factors are age, male gender and family history. People with a first-degree relative with Paget’s disease have a seven-fold increased risk of developing the disease when compared with controls.3 In the UK, around 15% of patients have a positive family history4 but this increases to around 40% in the Canadian province of Quebec through a founder effect.5
In some families, Paget’s disease is inherited as an autosomal dominant trait, most commonly due to mutations in the SQSTM1 gene but many other gene variants have been identified that predispose to Paget’s disease by genome wide association studies and family based studies.6
A reduction in prevalence and severity of Paget’s disease has been observed over the past 50 years and this is thought to be due to changes in environmental triggers for the disease. The nature of these is unclear at present but many potential triggers have been suggested including: viral infections, low dietary calcium intake during childhood, vitamin deficiency during skeletal growth and skeletal trauma.1,7
Clinical presentation
Many patients with Paget’s disease do not experience symptoms and epidemiological studies have estimated that only 7-15% of people with X-ray evidence of Paget’s disease come to medical attention.2,8
The most common presenting features in those that do present clinically are bone pain (52.2%), bone deformity (21.5%), deafness (8.9%) and pathological fractures (8.5%).8 In around 20% of patients, Paget’s disease is picked up as an incidental finding on blood tests or imaging carried out for another reason.8
Rarely, Paget’s disease may present with osteosarcoma (0.3% of patients) which usually manifests with a local increase in swelling and pain at an affected site, most commonly the pelvis, femur or humerus. Less common complications of Paget’s disease include: spinal stenosis, paraplegia due to spinal involvement, and cranial nerve compression; rare complications include giant cell tumours, high output cardiac failure and hypercalcaemia if the patient is immobilised, as illustrated in figure 1.
History and examination
On history taking it is important to establish if the presenting symptoms may be caused by Paget’s disease or an alternative diagnosis. The character of bone pain can vary between cases; some patients experience pain at rest which is dull and localised to an affected site while others can have pain which is worse with weight bearing or more localised because of pseudofractures.
It is also important to remember that in patients with Paget’s disease, symptoms such as pain can occur for other reasons and it is therefore vital to ascertain if the pain is localised to an affected site before attributing it to Paget’s disease. If it is not, other potential causes of pain should be considered, the most common of which is osteoarthritis.
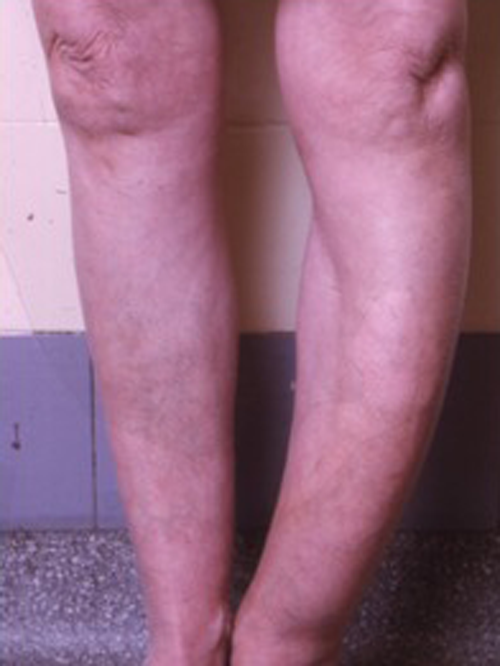
Deformity caused by Paget’s disease can usually be detected when the tibia (see figure 2) and skull are involved but deformity of other bones may also occur. Deafness occurs in around 20% of people with Paget’s disease who have skull involvement, mainly due to sensorineural problems.
Investigations
Diagnosis can be confirmed by X-ray which shows the characteristic features of osteolysis alternating with osteosclerosis, cortical thickening, bone expansion and bone deformity (see figure 3A). Radionuclide bone scan is the most sensitive way of detecting Paget’s disease (see figure 3B), but this is frequently not available to GPs.
If bone scans are not readily accessible, and Paget’s disease is suspected, plain X-rays of the abdomen (including the ribs and femoral heads), both tibias and the skull (including facial bones) should be performed. This approach has been found to detect Paget’s disease in 93% of cases.9
Routine biochemistry can also be helpful in the diagnosis of Paget’s disease that is metabolically active.
The typical abnormality is an isolated elevation in serum total alkaline phosphatase (ALP) in the presence of otherwise normal liver function tests. However, a normal ALP does not exclude the presence of Paget’s disease; when the disease affects a single bone, ALP values can be normal.
Serum calcium, phosphate, urea and electrolytes (U&E) and parathyroid hormone are usually normal in Paget’s disease, but 25(OH)D levels may be low reflecting the fact that the disease tends to affect older people in whom vitamin D deficiency is common.10
The main purpose of imaging is to identify which bones are affected to help determine if symptoms are due to Paget’s disease or another cause. Biochemical testing aims to determine if there is any evidence of metabolic activity. Magnetic resonance imaging (MRI) is not recommended for the diagnosis of Paget’s disease but can be valuable for the assessment of disease complications such as spinal stenosis in secondary care.
Referral
A suggested pathway for the investigation and referral of patients with suspected Paget’s disease is shown in figure 4. Patients who have bone deformity or symptoms that might be due to Paget’s disease should be referred to secondary care for further evaluation to determine whether treatment is required. In the UK, Paget’s disease is mostly dealt with by rheumatologists but in some areas, it will be the responsibility of endocrinologists or clinical biochemists.
Not all patients with Paget’s disease require referral to secondary care. In elderly patients who are asymptomatic, where the disease is picked up as an incidental finding, onward referral may not be necessary. There is no evidence as yet, to suggest that treatment is beneficial to patients without symptoms. If there is any doubt however, secondary care specialists will be happy to advise on whether referral is appropriate.
Management
Management of Paget’s disease is generally initiated in secondary care.
The main indication for medical treatment is bone pain localised to an affected site where there is evidence of increased metabolic activity.
Bisphosphonates are the treatment of choice and three are currently licensed for the treatment of Paget’s disease in the UK. These are: risedronate which is given in a dose of 30 mg daily by mouth for 2 months; zoledronic acid as a single intravenous infusion of 5 mg and pamidronate as three intravenous infusions of 60 mg on separate days.
Courses of treatment can be repeated as necessary if symptoms recur with evidence of increased metabolic activity, reflected by a raised ALP or increased uptake on bone scan. Vitamin D deficiency should be corrected prior to initiating bisphosphonate therapy to reduce the risk of hypocalcaemia which can be a complication, especially with intravenous therapy.11
Of these three agents, zoledronic acid is generally considered the first choice since it is most likely to give a favourable pain response and has the greatest inhibitory effects on elevated bone turnover.12 Risedronate and pamidronate can also help symptoms related to elevated bone turnover for 1-2 years or sometimes longer, but zoledronic acid has an even longer duration of action, lasting up to 5 years or more.13
All three bisphosphonates are generally well tolerated. A transient
flu-like illness is the most common adverse effect with intravenous therapy occurring in up to 50% of patients treated with zoledronic acid. It is usually mild but in about 10% may be more severe, requiring treatment with paracetamol or an NSAID. Upper gastrointestinal upset is the most common adverse effect with oral risedronate. Bisphosphonates are cleared by the kidney and are contraindicated if the eGFR is < 35 (zoledronic acid) or < 30 (risedronate and pamidronate).
Rare class adverse effects of bisphosphonates include uveitis, atypical femoral fractures (AFF) and osteonecrosis of the jaw (ONJ). Both AFF and ONJ are extremely rare in Paget’s disease since the doses given are generally much smaller than those required in osteoporosis or cancer-associated bone disease.12,14
Patients with coexisting disease such as osteoarthritis or nerve compression syndromes may also require additional therapy with analgesics, antineuropathic agents or NSAIDs to control pain fully.15
Calcitonin may be considered for the management of bone pain where bisphosphonates are contraindicated but long-term use has been associated with an increased risk of certain cancers. The usual dose is 100 units given three times weekly by subcutaneous injection for up to 3 months.
Denosumab is an osteoclast inhibitor that has also been used off label. It has been found to improve pain and reduce bone turnover but is reserved for use as a second-line agent in patients where bisphosphonates are contraindicated.16
Orthopaedic surgery is required in around 10% of patients with Paget’s disease. The most common reasons are fracture repair, hip or knee arthroplasty or spinal surgery to correct spinal stenosis.17 Surgery can be technically difficult in some cases due to bone deformity and osteosclerosis but the benefits almost always outweigh the risks and arthroplasty can produce excellent results.1
Other approaches such as physiotherapy and occupational therapy have not been widely studied but clinical experience suggests that the input of allied healthcare professionals can be helpful. In the PRISM study population, about 60% of individuals used aids and analysis of health assessment questionnaire data indicated that this significantly improved health related quality of life.18
Monitoring
It is usual to monitor the effects of bisphosphonate treatment both clinically and by serial ALP measurements. Typically, this will be done by measuring ALP and U&E at baseline and between 3 and 6 months after a course of treatment and then annually. If there is a recurrence of bone pain, localised to an affected site and ALP values become elevated, then this may indicate the need for further bisphosphonate treatment.
Not infrequently, pain may recur in the absence of an elevation in ALP. Under these circumstances the patient should be referred back to secondary care for evaluation. Often the specialist may consider a repeat bone scan to check if there is increased uptake at the painful site and if there is, another course of treatment may be given. If there is no increase in tracer uptake another cause may be sought. Patients should be advised to report any change or development of symptoms and a diagnosis of Paget’s should be noted in future consultations.
Future prospects
Many specialists prescribe bisphosphonates with the aim of suppressing markers of bone turnover in the hope that this will prevent
long-term complications but there is currently no evidence that this is beneficial.1 Indeed, clinical trials have shown no evidence that a treat to target approach is beneficial and some evidence suggests that it may be harmful compared with a symptom directed strategy.15,17
Research is currently in progress to determine if intervention with zoledronic acid is beneficial at the very early stages of the disease within the context of the ZiPP study19,20 which is due to report within the next 12 months. This multinational study included 222 people with a family history of Paget’s disease who had SQSTM1 mutations but who had not been diagnosed with the disease. Participants were recruited from centres in the UK, Spain, Italy, Belgium, Australia and New Zealand. Depending on the results this may herald a new approach to the treatment of Paget’s disease by coupling genetic testing for susceptibility with targeted intervention for those with very early disease.
REFERENCES
1 Ralston SH, Corral-Gudino L, Cooper C et al. Diagnosis and management of Paget’s disease of bone in adults: a clinical guideline. J Bone Miner Res 2019;34:579-604
2 van Staa TP, Selby P, Leufkens HG et al. Incidence and natural history of Paget’s disease of bone in England and Wales. J Bone Miner Res 2002;17:465-71
3 Siris ES, Ottman R, Flaster E, Kelsey JL. Familial aggregation of Paget’s disease of bone. J Bone Miner Res 1991;6:495-500
4 Visconti MR, Langston AL, Alonso N et al. Mutations of SQSTM1 are associated with severity and clinical outcome in Paget’s disease of bone. J Bone Miner Res 2010;25:2368-73
5 Morissette J, Laurin N, Brown JP. Sequestosome 1: mutation frequencies, haplotypes, and phenotypes in familial Paget’s disease of bone. J Bone Miner Res 2006;21 Suppl 2:38-44
6 Gennari L, Rendina D, Falchetti A, Merlotti D. Paget’s disease of bone. Calcif Tissue Int 2019;104:483-500
7 Siris ES. Epidemiological aspects of Paget's disease: family history and relationship to other medical conditions. Semin Arth Rheum 1994;23:222-25
8 Tan A, Ralston SH. Clinical presentation of Paget’s disease: evaluation of a contemporary cohort and systematic review. Calcif Tissue Int 2014;95:385-92
9 Guañabens N, Rotés D, Holgado S et al. Implications of a new radiological approach for the assessment of Paget disease. Calcif Tissue Int 2012;91:409-15
10 Rendina D, De Filippo G, Merlotti D et al. Vitamin D status in Paget disease of bone and efficacy-safety profile of cholecalciferol treatment in pagetic patients with hypovitaminosis D. Calcif Tissue Int 2019;105:412-22
11 Merlotti D, Rendina D, Muscariello R et al. Preventive role of vitamin D supplementation for acute phase reaction after bisphosphonate infusion in Paget’s disease. J Clin Endocrinol Metab 2020;105
12 Corral-Gudino L, Tan AJ, Del Pino-Montes J, Ralston SH. Bisphosphonates for Paget’s disease of bone in adults. Cochrane Database Syst Rev 2017;12:CD004956
13 Reid IR, Lyles K, Su G et al. A single infusion of zoledronic acid produces sustained remissions in Paget disease: data to 6.5 years. J Bone Miner Res 2011;26:2261-70
14 Ralston SH. Bisphosphonates in the management of Paget’s disease. Bone 2020;138:115465
15 Langston AL, Campbell MK, Fraser WD et al. Randomised trial of intensive bisphosphonate treatment versus symptomatic management in Paget’s disease of bone. J Bone Miner Res 2010;25:20-31
16 Reid IR, Sharma S, Kalluru R, Eagleton C. Treatment of Paget’s disease of bone with denosumab: case report and literature review. Calcif Tissue Int 2016;99:322-25
17 Tan A, Goodman K, Walker A et al. Group P-ET. Long-term randomized trial of intensive versus symptomatic management in Paget’s disease of bone: The PRISM-EZ study. J Bone Miner Res 2017;32:1165-73
18 Langston AL, Campbell MK, Fraser WD et al. Clinical determinants of quality of life in Paget’s disease of bone. Calcif Tissue Int 2007;80:1-9
19 Cronin O, Subedi D, Forsyth L et al. Characteristics of early Paget’s disease in SQSTM1 mutation carriers: baseline analysis of the ZiPP study cohort. J Bone Miner Res 2020;35:1246-52
20 Cronin O, Forsyth L, Goodman K et al. Zoledronate in the prevention of Paget’s (ZiPP): protocol for a randomised trial of genetic testing and targeted zoledronic acid therapy to prevent SQSTM1-mediated Paget’s disease of bone. BMJ Open 2019;9:e030689