SEARCH
NEUROLOGY
Diagnosis and management of epilepsy in adults
26 Sep 2022
According to the International League Against Epilepsy, epilepsy can be diagnosed if any of the following criteria are met: at least two unprovoked seizures occurring on separate days (seizures within 24 hours count as one event); one unprovoked seizure with at least a 60% risk of recurrence over the next ten years on the basis of associated clinical factors (such as a recent stroke or brain tumour); diagnosis of a specific epilepsy syndrome. Convulsive events should be described rather than given a label. The EEG can establish the diagnosis of epilepsy and distinguish between focal and primary generalised epilepsy. An MRI brain scan is usually mandatory.
 |
|GPs should be vigilant for Parkinson’s disease
25 Sep 2021
Parkinson's disease is the second most common neurodegenerative disease. Parkinsonism is defined as bradykinesia in combination with at least one of rest tremor or rigidity. Parkinson's disease is a clinical diagnosis. Examination should include gait and posture, decreased blink rate and a lack of spontaneous facial movements. Speech will be hypophonic. Evidence of a resting tremor should be sought and the patient assessed for joint rigidity and bradykinesia. After initial assessment, prompt referral to a neurologist is important to confirm the diagnosis and discuss management.
 |
|History and examination pivotal in diagnosis of Bell’s palsy
24 Sep 2021
Bell’s palsy is the most common cause of facial palsy and presents as a unilateral lower motor neurone facial weakness in association with sensory and parasympathetic dysfunction. Maximal facial weakness occurs within 72 hours and most cases recover over the following few weeks. It is essential to consider alternative causes of acute peripheral facial palsy including: infection, malignancy and autoimmune disease. Urgent referral to neurology or ENT, depending on local referral pathways, is warranted if there is uncertainty about the diagnosis or there are atypical features.
 |
|GPs should be vigilant for acute deterioration in myasthenia gravis
24 Sep 2020
Myasthenia gravis is an autoimmune disorder of neuromuscular junction transmission. It is relatively rare, with an approximate annual incidence of 1 per 100,000 population, and prevalence of 15 per 100,000 population in the UK. An ocular presentation may include fatiguing ptosis or diplopia. Typically, symptoms ‘fatigue’ (the physical power of the muscle deteriorates rapidly with repeated activity) and become more noticeable as the day progresses. More generalised symptoms include fatiguing difficulty with speech or swallowing. There may be fatiguing weakness of the arms and legs. The diagnosis will usually be confirmed by referral to a neurologist.
Detailed history the cornerstone of epilepsy diagnosis
24 Sep 2020
The incidence of epilepsy in the UK is estimated to be 50 per 100,000 per year and up to 1% of the population have active epilepsy. The diagnosis of epilepsy will usually be made in a neurology clinic. A generalised seizure as part of a generalised epilepsy syndrome may occur without warning but may be preceded by blank spells or myoclonic jerks. A generalised seizure with focal onset may be preceded by an aura. Brain imaging is required in almost all cases where epilepsy is suspected, the only possible exception being people with generalised epilepsies proven on EEG. MRI is the imaging modality of choice.
Optimising the management of neuropathic pain
25 Sep 2019
Neuropathic pain is defined as ‘pain that is caused by a lesion or disease of the somatosensory nervous system.’ The International Association for the Study of Pain (IASP) pain grading system is a simple way of determining the likelihood of a neuropathic component. If the history suggests a relevant neurological lesion or disease and the patient describes pain in an anatomically plausible distribution of a nerve, neuropathic pain is ‘possible’; it is ‘probable’ if there are corresponding examination findings in that same distribution and ‘definite’ if there is a confirmatory diagnostic test.
 |
|Tackling medication overuse headache in primary care
24 Sep 2018
Medication overuse headache (MOH) occurs as a complication of the management of primary headache disorders, mainly migraine and tension type headache. MOH does not occur in cluster headache unless there is associated migraine. MOH is defined as headache occurring on 15 or more days per month, that has evolved in association with the frequent use of acute medication over a period of more than 3 months. Any medication used for the acute treatment of headache can cause MOH.
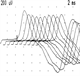
Diagnosis and management of sleep-related epilepsy in adults
24 Sep 2018
Nocturnal epilepsies account for 10-15% of all epilepsies, and 80% of nocturnal epilepsies in adults are focal. They present a diagnostic challenge as they can be difficult to differentiate from normal movements and behaviour during sleep and also from several non-epileptic, sleep-related, motor and behavioural disorders. More than 90% of the seizures in sleep-related hypermotor epilepsy (SHE) occur during sleep. Seizures in SHE are simple partial seizures which easily wake the patient so that they will usually be able to recall the seizures, often describing auras of somatic sensations or feeling unable to breathe. SHE seizures have a rapid onset and offset, a short duration (usually < 2 min), and a stereotyped motor pattern for that individual.
Diagnosis and management of complex regional pain syndrome
22 Sep 2017
Complex regional pain syndrome (CRPS) is a chronic debilitating painful condition comprising unremitting pain, sensory, sudomotor, vasomotor and motor abnormalities in the affected extremity. It has a peak incidence in the 55-75 age group and there is an association with asthma and migraine. CRPS is three times more common in women than men. CRPS should be suspected in any patient presenting with persistent pain in an extremity beyond the expected period of tissue healing following an acute injury, sprain, fracture or surgical procedure. Severe pain in a glove or stocking distribution is the predominant symptom in > 90% of cases.
Identifying neurological causes of daytime sleepiness
22 Sep 2017
The prevalence of sleep complaints in adults in a primary care setting is > 10%. The most frequently seen condition by far is that of primary insomnia, which affects 10% of adults on a chronic basis. In contrast to primary insomnia, in which most patients report tiredness and fatigue during the day but are unable to sleep during the day either, the second most frequent sleep disorder encountered, obstructive sleep apnoea, is typified by excessive daytime sleepiness. Patients with primary insomnia or fatigue syndromes typically will score low on the Epworth Sleepiness Scale (ESS < 3) whereas those with organic sleep pathologies or sleep restriction will score higher. A score > 10 is seen as 'pathological', with a mean ESS in the population of 5-6.
 |
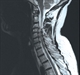
|Diagnosis and management of motor neurone disease
23 Sep 2016
Motor neurone disease is a rapidly progressive and fatal neurodegenerative condition which causes progressive weakness, with normal sensation. Key clinical presentations include bulbar (slurred or difficult speech, problems swallowing, tongue fasciculation), limb (typically in one limb with weakness and muscle wasting), respiratory (breathlessness, chest muscle fasciculation) and cognitive features (behavioural change, emotional lability, features of frontotemporal dementia).
 |
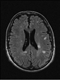
|Tailor treatment to the patient with neuropathic pain
23 Sep 2016
Neuropathic pain is defined as pain that is caused by a lesion or disease of the somatosensory nervous system and is estimated to affect 6-8% of the general population. A low threshold of suspicion in conditions associated with neuropathic pain can aid diagnosis. Typical neuropathic descriptors include burning, shooting, electric shock pain with numbness, pins and needles or itching.
 |
|Early accurate diagnosis crucial in multiple sclerosis
24 Sep 2015
In around 85% of cases, multiple sclerosis (MS) starts with an acute neurological episode, a clinically isolated syndrome, which is considered to be the first clinical episode of relapsing-remitting MS. It is characterised by the presence of acute relapses, after which there is normally good functional recovery. Investigations need to rule out conditions that can mimic an inflammatory-demyelinating disease of the CNS and determine the presence of dissemination in space and dissemination in time of the inflammatory-demyelinating disease. There is no confirmatory test for MS and it remains essentially a clinical diagnosis.
 |
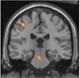
|Seizure classification key to epilepsy management
24 Sep 2015
The diagnosis of epilepsy is often incorrect, potentially in up to 20% of cases, so should be revisited if seizures are not responding to treatment. SIGN recommends that the diagnosis of epilepsy should be made by an epilepsy specialist, ideally in the setting of a dedicated first seizure or epilepsy clinic. Diagnosis relies primarily on the history. Distinguishing between a genetic generalised epilepsy and a focal epilepsy is vital as this influences investigation, treatment and prognosis.
 |
|Migraine is underdiagnosed and undertreated
23 Sep 2014
Migraine is a common neurovascular disorder characterised by attacks of head pain that are typically unilateral and often described as severe and throbbing in association with nausea and sensitivity to sensory input, i.e. light, sound and head movement. NICE guidelines recommend adopting the stepped-down approach to management. They suggest a combination of a triptan, NSAID or paracetamol, and an anti-emetic taken as early as possible during the headache. [With external links to the evidence base]
 |
|Improving outcomes for chronic pain in primary care
23 Sep 2014
Although the patient’s goal is often complete pain relief, this is rarely a realistic outcome, so the role of the physician in managing chronic pain involves optimising pain relief as far as possible. A thorough biopsychosocial assessment is essential so that an individualised multidisciplinary approach to management can be developed. The aims of assessment of chronic pain are to rule out any underlying serious pathology, identify the pain mechanism and identify and evaluate risk factors that contribute to chronicity.
 |
|Managing patients with cluster headache in primary care
23 Sep 2013
Cluster headache is a strictly unilateral headache that occurs in association with cranial autonomic features. The diagnosis is purely clinical and it is therefore crucial to take a good history looking for its distinctive features. Specialist advice should be sought: at first presentation for confirmation of diagnosis, and development of a plan for managing current and future cluster bouts; and where first-line treatments fail.
 |
|Community stroke rehabilitation helps patients return to work
23 Sep 2013
A quarter of stroke survivors are under the age of 65 meaning that many are in work and/or have responsibility for caring for children or elderly parents. With a comprehensive rehabilitation team, patients with more complex or severe disability can be rehabilitated in the community providing that the home environment can be suitably adapted. The core components of a community-based programme can be broadly defined as improving emotional wellbeing, communication, cognitive function and physical independence and supporting return to work.
Diagnosing and managing muscular dystrophy
20 Sep 2012
Muscular dystrophy refers to a range of muscle diseases caused by defects in muscle proteins, leading to death of the muscle cells, with loss of muscle tissue, and weakness. Muscular dystrophy may present at any age from perinatal to old age. The muscular dystrophies all have a genetic basis, although not all causes have been identified. Patients may present in primary care undiagnosed, or with a diagnosis and established disability when transferring practice. The development of clinical symptoms is usually gradual, and the earliest features may be difficult to identify and determine. With established disease the presence of muscle weakness and wasting is clear. In children, the presentation may be delayed walking or poor performance in sporting activity. In children and adults presenting symptoms may include: difficulty raising from a squat;difficulty raising from a chair; difficulty lifting arms above the head; poor balance; drooping eyelids; joint contractures.
 |
|Rapid diagnosis of TIA reduces risk of subsequent stroke
19 Sep 2012
A transient ischaemic attack (TIA) is defined as a stroke that recovers completely within 24 hours of onset. TIA and stroke are the same disease and both need to be treated with equal urgency. Patients with ongoing symptoms and signs at the time of assessment, however early after onset and even if improving, need to be treated as stroke with rapid transfer to an inpatient stroke service.
 |
|GPs have key role in managing motor neurone disease
21 Sep 2011
Motor neurone disease (MND) is a rapidly progressive neurodegenerative condition. It affects people of all ages, but is more common with increasing age (especially over 50 years) and men are affected twice as often as women. The prevalence is relatively low, as individuals usually live for only a few years with MND. There are around 1,200 new diagnoses a year, and 4,000 people affected at any one time in the UK. It will probably become more frequent with an ageing population, and the prevention of death from vascular and other causes. The Motor Neurone Disease Association has a network of regional care advisers, and visitors, and can provide support and information for the GP. MND is a devastating physical condition, and can have equally devastating effects on emotions, also affecting family, friends, and members of the multidisciplinary team. Some patients experience difficulties accessing resources, which the GP may help in mobilising. There may also be problems in understanding and communication, where the GP may play a key role.
 |
|Evaluating first seizures in adults in primary care
20 Sep 2011
Many patients experiencing a first seizure will present to their GP. A seizure is defined as the clinical manifestation resulting from an excessive and abnormal discharge of a population of neurones. The individual lifetime risk of developing a non-febrile seizure is around 5% and approximately a third of patients will go on to experience further seizures and hence be diagnosed as having epilepsy. When evaluating a patient with a first seizure an accurate history is key and other potentially life-threatening diagnoses, especially cardiac disease, must be ruled out. Patients who have fully recovered should be directly referred to a first seizure clinic, where further tests and AEDs may be considered. Doctors should provide support and advice to the first seizure patient and be aware of the medical and social consequences of a diagnosis of seizures or epilepsy. Patients who have not recovered from their seizure, have an abnormal examination, or about whom there is diagnostic doubt, should be referred to the emergency department or specialist services for further investigations including brain imaging and lumbar puncture.
 |
|History central to diagnosing myasthenia gravis
21 Sep 2010
MG has a prevalence of around 20 per 100,000, so an average UK practice may expect to have only one patient. The condition is treatable. The incidence is bimodal with a 2:1 female to male ratio in the younger population and a reversed sex ratio over the age of 60. The reported incidence has been increasing since the mid-1980s, mainly because of increased recognition of late-onset disease. However, the condition is probably still underrecognised in the very elderly and may be mistaken for other disorders such as stroke, motor neurone disease or Parkinson's disease.
 |
|Systematic approach needed to establish cause of vertigo
20 Sep 2010
Vertigo is a common but complex challenge. In one postal survey of patients from general practice, 7% reported at least one episode of vertigo in the past year. A full-time GP may expect to encounter 10-20 cases of vertigo each year and it is often seen as a potentially difficult symptom to diagnose and manage. The first problem is that the terms vertigo, dizziness, giddiness and imbalance are used in different ways by different clinicians. Dizziness and vertigo are often used interchangeably by both patients and clinicians (including the authors). The second problem is that the control of balance is complicated and diagnosing the cause of a balance disorder can take time which is usually at a premium in most clinics. Third, vertigo can be caused by many different pathologies, some of which are potentially life threatening, and it is managed by several specialties. However, much of this can be overcome by using a logical, systematic approach to the diagnosis and subsequent referral or management of the patient.